Agricultural Sciences, 2015, 6, 630-662 © 2015 by authors and Scientific Research Publishing Inc.
DOI: 10.4236/as.2015.67062
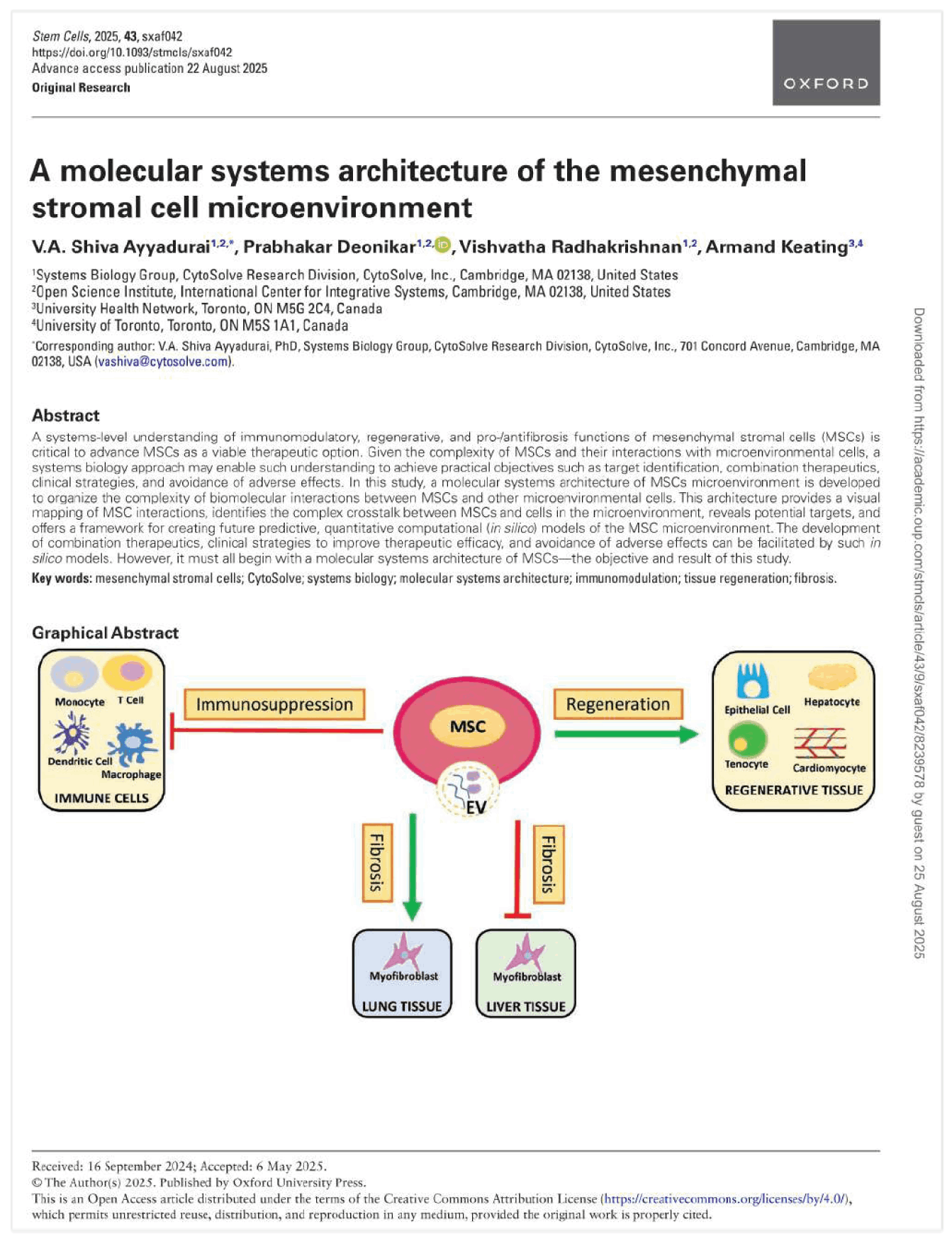
Abstract:
Safety assessment of genetically modified organisms (GMOs) is a contentious topic. Proponents of GMOs assert that GMOs are safe since the FDA’s policy of substantial equivalence considers GMOs “equivalent” to their non-GMO counterparts, and argue that genetic modification (GM) is simply an extension of a “natural” process of plant breeding, a form of “genetic modification”, though done over longer time scales. Anti-GMO activists counter that GMOs are unsafe since substantial equivalence is unscientific and outdated since it originates in the 1970s to assess safety of medical devices, which are not comparable to the complexity of biological systems, and contend that targeted GM is not plant breeding. The heart of the debate appears to be on the methodology used to determine criteria for substantial equivalence. Systems biology, which aims to understand complexity of the whole organism, as a system, rather than just studying its parts in a reductionist manner, may provide a framework to determine appropriate criteria, as it recognizes that GM, small or large, may affect emergent properties of the whole system. Herein, a promising computational systems biology method couples known perturbations on five biomolecules caused by the CP4 EPSPS GM of Glycine max L. (soybean), with an integrative model of C1 metabolism and oxidative stress (two molecular systems critical to plant function). The results predict significant accumulation of formaldehyde and concomitant depletion of glutathione in the GMO, suggesting how a “small” and single GM creates “large” and systemic perturbations to molecular systems equilibria. Regulatory agencies, currently reviewing rules for GMO safety, may wish to adopt a systems biology approach using a combination of in silico, computational methods used herein, and subsequent targeted experimental in vitro and in vivo designs, to develop a systems understanding of “equivalence” using biomarkers, such as formaldehyde and glutathione, which predict metabolic disruptions, towards modernizing the safety assessment of GMOs.